
- 解剖・生理
- 動脈血栓と静脈血栓
- 汎血球減少
- 検査
- 貧血の鑑別(必ずMCVを計算する!!)
- 赤血球系の疾患
- 止血・凝固系の疾患
- DIC:Disseminated intravascular coagulation(播種性血管内凝固症候群)
- 造血器腫瘍
- 造血幹細胞移植 Hematopoietic stem cell transplantation
解剖・生理
血液は体重の1/13(8%)で、全血液量の1/3を失うと生命に危険が及ぶ!
造血部位
| 造血部位 | ||
| 胎生期 | 卵黄嚢→肝臓>脾臓 | 胎生第2週から卵黄嚢→第6週から主に肝臓(髄外造血) |
| 出生後 | 骨髄(全ての骨) | 造血機能を有する骨髄=赤色骨髄 |
| 成人 | 骨髄(体幹部の骨) | 造血機能を有しない骨髄=黄色骨髄(脂肪に置換) |
| 髄外造血 | 脾臓>肝臓 | 骨髄線維症、癌の骨髄転移、重篤な慢性貧血 |
胎児血中には卵黄嚢や肝細胞で産生されたAFPが存在する。
RBCは新生児に600万と多血気味だが、10歳頃に成人値となる。
WBC系は出生直後17000と高値を示すが、生後1〜2週間で著減し、以降は成人値となる。
造血幹細胞の特徴
| ①自己複製能 | IL-3、SCFによって自己複製していると考えられている。 |
| ②多分化能 | 骨髄微小環境(ニッチ)によって分化増殖すると考えられている。 |
| ③CD34発現 | G-CSF投与すると多分化能を維持したCD34陽性細胞が末梢血に現れる。 |
各血球の寿命・特徴
| 寿命 | 特徴 | 分化増殖因子 | |
| 好中球 | 数日 | 桿状核→分葉核に分化する。細胞質のアズール顆粒(リソソーム酵素、MPO、エラスターゼ)や特殊顆粒(アルカリホスファターゼ、リゾチーム)が黄褐色に染色される。 | G-CSF |
| 好酸球 | 数日 | 細胞質が赤く染色される。核が2つ。ロイコトリエンやPAFを含む顆粒を持つ。 | IL-5 |
| 好塩基球 | 数日 | 細胞質が青く染色される。ヒスタミンやヘパリンを含む顆粒を持つ。 | IL-5 |
| リンパ球 | 数日〜1年 | 核の割合が大きい。2/3がT細胞、1/3がB細胞、数%がNK細胞。 | IL-2など |
| 単球 | 数日 | 白血球の中で一番大きく、核に切れ目がありハート型に見えるかも。 | M-CSF |
| 血小板 | 1〜2週間 | 巨核球の細胞質がちぎれて血小板となる。 | TPO |
| 赤血球 | 約120日 (4ヶ月) |
赤芽球から脱核して網赤血球になると末梢血に放出され、1〜2日後に成熟赤血球になる。 | EPO |
赤血球の合成・分解
酸素解離曲線は呼吸器総論を参照。
| ヘム合成 | 【赤芽球中】グリシン+サクシニルCoA→δ-アミノレブリン酸→プロトポルフィリン+鉄→ヘム+グロビン→ヘモグロビン(Hb) |
| ヘム分解 | 網内系(脾臓、骨髄など)でマクロファージに貪食→Hb→ヘム+グロビン→鉄(再利用)+ビリベルジン→間接ビリルビン→アルブミンと結合して肝臓に輸送され、水溶性の直接ビリルビン→胆汁と共に十二指腸に排泄 (ビリルビンは腸管でウロビリノゲンとなり回腸で再吸収され腎臓から排泄される) |
| 鉄の吸収 | 食物中のFe3+が胃酸やVCで還元されFe2+となり十二指腸から吸収される(約1mg/日)。血中に取り込まれた鉄は鉄輸送蛋白のトランスフェリン(Tf)と結合し血清鉄となる。血清鉄は骨髄に移動して赤芽球に取り込まれる。 |
| 鉄の保存 | 血中に過剰になった血清鉄(Fe+Tf)は肝臓に運ばれ、鉄はアポフェリチンと結合してフェリチン(貯蔵鉄)となる。体内に鉄は約3g存在し、1/3は貯蔵鉄、2/3は血清鉄として存在する。フェリチンが変性すると不溶性のヘモジデリンとなる。 |
| 鉄の排泄 | 表皮細胞、粘膜上皮細胞の脱落や汗により排泄される(約1mg/日)。ただし、月経時には15〜40mg/日失われる。 |
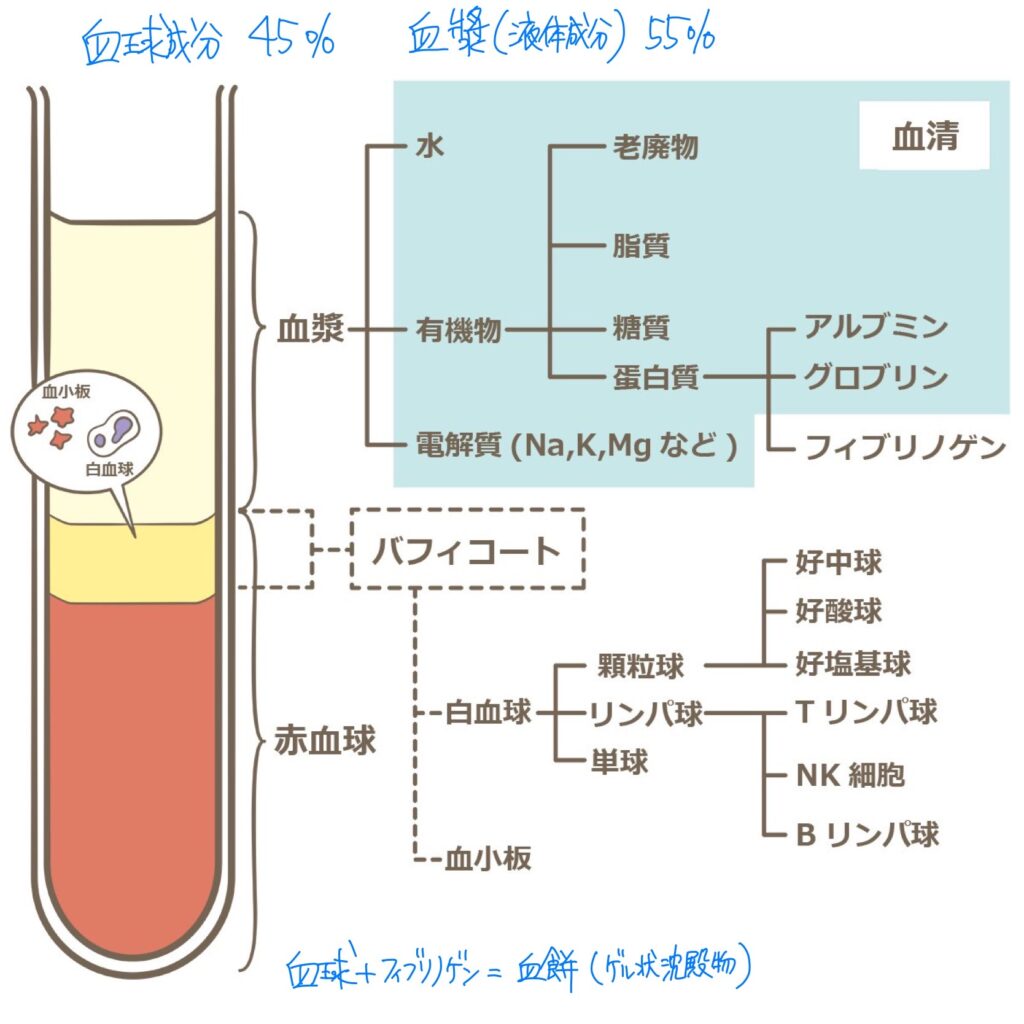
血小板・止血機構
血小板は1/3が脾臓に貯蔵、2/3が血中に存在する。血小板数↓により出血時間が延長する。
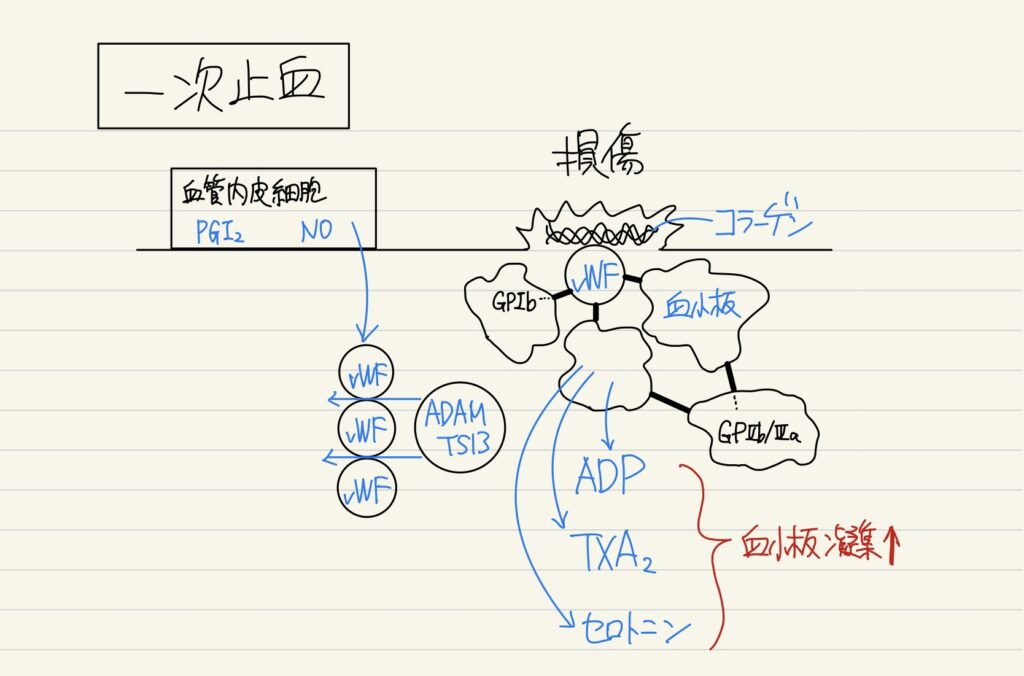
【一次止血】
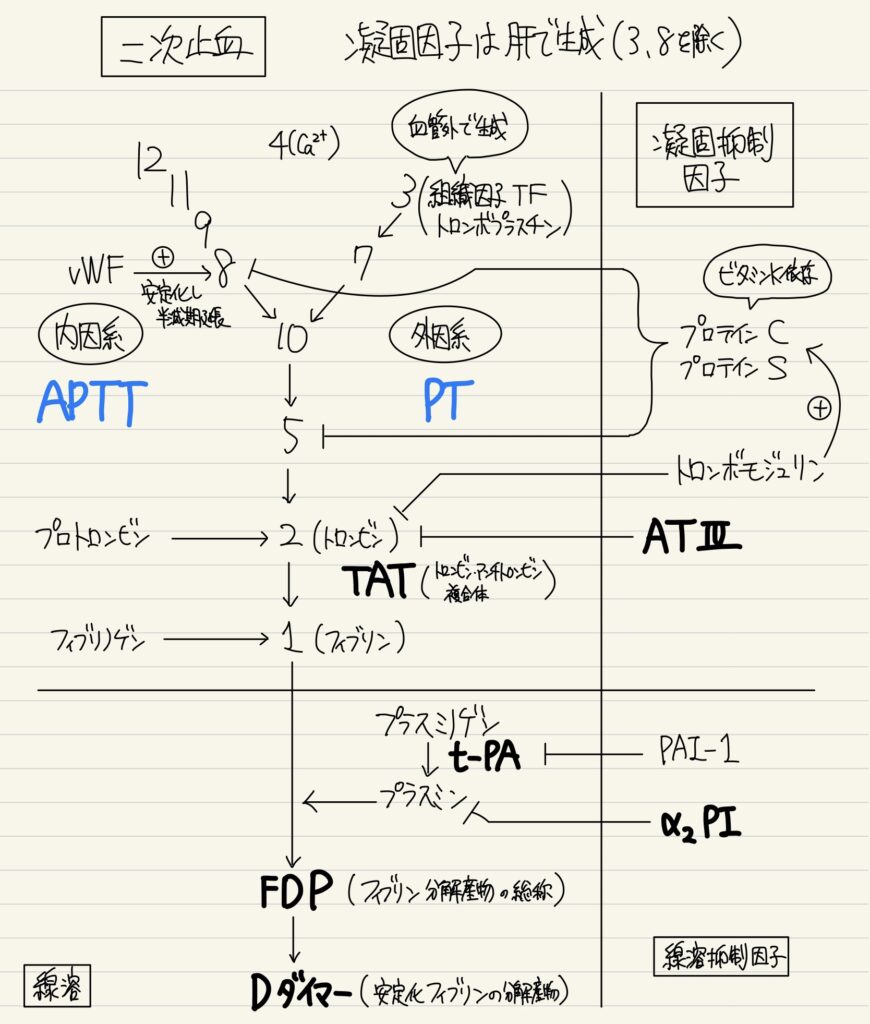
【二次止血】
動脈血栓と静脈血栓
| 動脈血栓 | 静脈血栓 | |
| 機序 | 動脈硬化を背景に血管内皮細胞が障害され、その部分に血小板が凝集し血栓が形成される。 | 血液が鬱滞し、赤血球にフィブリン血栓が形成される。 |
| 疾患 | 虚血性心疾患、TIA アテローム血栓性脳梗塞 末梢動脈疾患 |
静脈血栓塞栓症 |
| 治療 | 抗血小板 | 抗凝固薬 |
汎血球減少
汎血球減少症する疾患→最強SMAP丼:再生不良性貧血、巨赤芽球性貧血、SLE、MDS、Acute(急性白血病)、PNH、血球貪食症候群
| 骨髄での血球産生低下 | 末梢血での血球破壊亢進 |
| 再生不良性貧血(先天性:Fanconi貧血) 骨髄異形成症候群 巨赤芽球性貧血 急性白血病 多発性骨髄腫 PNH がんの骨髄転移 など |
脾機能亢進(門脈圧亢進) 膠原病(SLEなど) 血球貪食症候群 DIC など |
検査
NAPスコア
NAP(好中球アルカリホスファターゼ)は好中球の成熟度を表す。
顆粒の数や分布を点数化したものをNAPスコアと言う。
| NAP↑ | CMLの急性転化、真性赤血球増加症、原発性骨髄線維症、本態性血小板血症、類白血病反応、、化膿性感染症、妊娠 |
| NAP↓ | CMLの慢性期、PNH、一部の急性骨髄性白血病(M2)、骨髄異形性症候群 ナプキン無いと困るから夜のAM2時にこっそり行け:NAP無い、CML、発作性夜間Hb尿症、M2、骨髄異形性症候群 |
Coombs試験
| 検査方法 | 疾患 | |
| 直接クームス試験 | 患者赤血球に※特定の抗体を入れる。凝集すれば自己赤血球に対する抗体がある。 ※抗赤血球抗体=Coombs抗体 |
免疫性:90%がAIHA 新生児溶血性疾患 不適合輸液後 |
| 間接クームス試験 | 患者血清に赤血球と二次抗体(IgG)を入れる。凝集すれば非自己赤血球に対する抗体がある。 | 血液型の判定(ウラ試験) 輸血前のクロスマッチ(主試験) Rh不適合妊娠 |
貧血の鑑別(必ずMCVを計算する!!)
正球性〜大球性貧血の鑑別
※無効造血:骨髄内で溶血し消失するため末梢血に出現しない=網赤血球↓
| 網赤血球 | 汎血球 | EPO | キーワード | |
| 再生不良性貧血 | ↓ | 減少 | ↑ | フェリチン↑血清鉄↑ 【骨髄生検】骨髄低形成・脂肪髄 |
| 赤芽球癆 | ↓ | ↑ | 血清鉄↑フェリチン↑TIBC↓ | |
| 腎性貧血 | 慢性腎不全 | |||
| 骨髄線維症 | ↑ | 巨大脾腫 | ||
| MDS | 減少 | ↑ | 無効造血 【骨髄生検】骨髄過形成 |
|
| 多発性骨髄腫 | M蛋白↑、高Ca血症 | |||
| PNH | ↑ | 減少 | ||
| 巨赤芽球性貧血 | 減少 | 無効造血 大球性:MCV130以上 |
||
| 急性白血病 | 減少 |
溶血性貧血の鑑別
| 病態 | 【血管外溶血】先天性:HS、サラセミア、後天性:AIHA 【血管内溶血】先天性:G6PD欠損症、後天性:PNH、TTP、HUS、DIC、機械弁置換 |
| 症状 | 【共通】黄疸(眼球結膜黄染)、胆石 【血管外溶血】脾腫 【血管内溶血】脾腫なし、Hb尿、ヘモジデリン尿 |
| 検査 | 【尿検査】 尿中ウロビリノーゲン↑ 【血液検査】 溶血所見:LDH↑、間接Bil↑、ハプトグロビン↓、K↑、代償的に網赤血球↑ 【骨髄生検】 赤芽球過形成 |
| 治療 | 【血管外溶血】脾摘、免疫性ならステロイド投与 |
【鑑別】
| 症状 | 疾患 | 特徴 | ||
| 血管外溶血 | 脾腫 | 先天 | HS(膜異常) | 球状赤血球、直接Cooms試験(ー) |
| βサラセミア(血色素異常) | 標的赤血球、HbF・HbA2、無効造血 | |||
| 後天 | AIHA(抗体) | 直接Cooms試験(+)、Ⅱ型アレルギー、脾臓で溶血 | ||
| 寒冷凝集素症 | 肝臓で溶血 | |||
| 血管内溶血 | Hb尿 | 先天 | G6PD欠損症(赤血球膜の不安定化) | |
| 後天 | PNH(幹細胞突然変異) | 早朝褐色尿、汎血球減少、NAPスコア↓、CD55/59 | ||
| TTP(赤血球破砕) | 血小板減少、凝固能正常 | |||
| HUS(赤血球破砕) | ||||
| DIC(赤血球破砕) | ||||
| 機械弁置換(赤血球破砕) | ||||
| 特発性門脈亢進症(脾機能亢進) |
赤血球系の疾患
ヘモクロマトーシス
| 病態 | 組織に過度な鉄の蓄積が起こり、臓器障害をきたした状態。 鉄は能動的排泄はされないため、過剰投与によりヘモクロマトーシスを起こす。 特に肝臓、膵臓、皮膚、心臓、関節、甲状腺、下垂体、精巣などの諸臓器に過剰に沈着し、症状を引き起こす。 原発性:常染色体劣性遺伝で、日本ではまれ。 続発性:貧血に対する頻回の赤血球輸血(最多)、無効造血をきたす疾患など |
| 症状 | <四主徴>黒い頭皮が肝心! ①糖尿病 ②皮膚色素沈着:鉄沈着によって皮膚に暗褐色調の色調変化 ③肝硬変 ④心不全 |
| 検査 | 【血液検査】フェリチン著増↑ |
| 治療 | 鉄キレート療法、貧血を伴わない場合は瀉血 |
再生不良性貧血 AA:Aplastic Anemia
| 病態 | 何らかの免疫異常が起こりキラーT細胞が活性化され、骨髄系幹細胞が減少して骨髄の造血能低下により汎血球減少が生じる貧血。特発性には急性型と慢性型がある。 先天性:Fanconi貧血など 特発性:特発性(80%)、二次性(クロラムフェニコール、ベンゼン、肝炎ウイルス) |
| 症状 | 貧血共通症状、易感染性(発熱など)、出血傾向(四肢の紫斑=点状出血など) |
| 検査 | 【血液検査】 汎血球減少:WBC↓(相対的リンパ球増加)RBC↓網赤血球↓PLT↓代償的にEPO↑ 鉄が使われないため血清鉄↑フェリチン↑TIBC↓(肝にFeを運ぶ必要ないため) 【尿検査】EPO↑ 【骨髄生検】骨髄低形成(有核細胞↓、脂肪髄化) |
| 治療 | 【40歳未満かつ赤血球輸血を必要するstage3〜5】 造血幹細胞移植(40〜65歳でも治療無効例には考慮) 【上記に該当しない患者】 免疫抑制療法4剤:シクロスポリン+抗胸腺細胞グロブリン(ATG)+ステロイド(ATGによるアレルギー予防)+エルトロンボパグ(TPO受容体作動薬でRBC&PLT産生↑) あんたが捨てた白いパグ:ATG、ステロイド、シクロスポリン、エルトロンボパグ 支持療法:赤血球輸血(ヘモクロマトーシスに注意)、血小板輸血、G-CSF投与 |
赤芽球癆 PRCA:Pure Red Cell Aplasia
| 病態 | 骨髄で赤芽球のみが減少する疾患(再不RBCのみVer)。先天性と後天性がある。 <後天性> 急性:薬剤性もしくはパルボウイルスB19が赤芽球に感染して破壊するため、成人の場合は一過性(1〜3週間)の貧血を起こす。赤血球の寿命が短い溶血性貧血患者や胎児では症状が顕著となる。 慢性:自己免疫性に赤芽球が破壊される(胸腺腫(9%)を合併)。 |
| 症状 | ①貧血共通症状 ②溶血症状:眼球結膜黄染、脾腫(血管外) ③無形成発作:溶血性貧血患者や胎児にパルボウイルスB19感染すると貧血が一挙に悪化して重度の貧血となる。 |
| 検査 | 【血液検査】 RBC↓網赤血球↓、鉄が使われないため血清鉄↑フェリチン↑TIBC↓ 【骨髄生検】 赤芽球系のみ低形成 |
| 治療 | 急性:被疑薬中止、対症療法 慢性:シクロスポリン、ステロイド、胸腺腫合併の場合は摘出 |
温式自己免疫性溶血性貧血 AIHA:Autoimmune Hemolytic Anemia
| 疫学 | 若年女性と高齢者に好発(二相性)、後天性溶血性貧血で最多 |
| 病態 | 赤血球膜に対する自己抗体(IgG)が産生され、Ⅱ型アレルギーの機序で脾臓のマクロファージで貪食されて血管外溶血が起こる。約20%はITPを合併(Evans症候群)。 <原因> 特発性(半数):原因不明 二次性(半数):AIHAの分類を参照 |
| 症状 | ①貧血共通症状 ②溶血性貧血症状:黄疸、胆石 ③血管外溶血:脾腫 |
| 検査 | 【血液検査】 溶血共通:間接Bil↑LDH↑K↑ハプトグロビン↓網赤血球↑ 二次性の場合は直接Coombs試験(+)、ITP合併の場合は抗血小板抗体(+) 【末梢血塗抹標本】 小球性赤血球(温式AIHA)、大小不同のRBC |
| 治療 | ①ステロイド投与 ②抵抗性の場合は、脾摘 or 免疫抑制薬(シクロホスファミド、アザチオプリンなど) ③γ-グロブリン大量投与、蛋白同化ステロイド(造血促進作用)など ※冷式AIHAの場合は寒冷を避け保温 |
【AIHAの分類】
| 疾患 | 抗体 | 溶血部位 | 二次性 | |
| 温式 (37度) |
温式AIHA (90%以上) |
IgG | 血管外溶血 (脾で分解) |
SLE、RA、薬剤性、CLL、悪性リンパ腫 |
| 冷式 (0〜4度) |
寒冷凝集素症 (約8%)→中高年 |
IgM | 血管外溶血 (肝で分解) |
先行感染(マイコプラズマ、伝染性単核球症)、悪性リンパ腫 |
| 発作性寒冷Hb尿症 (約2%)→乳幼児 |
IgG | 血管内溶血 | 先行感染(梅毒、麻疹、水痘、伝染性単核球症) |
遺伝性球状赤血球症 HS:Hereditary Spherocytosis
| 疫学 | 多くは小児期に発症。先天性溶血性貧血で最多。 |
| 病態 | 常染色体優性遺伝(約2/3)により、赤血球膜蛋白異常が生じ、赤血球が球状化し、脾臓で血管外溶血が起こる。球状化するため正球性高色素性貧血となる(MCHC高値)。 HSは貧血が高度となることは比較的少なく、成人になって発見されることもある。 |
| 症状 | ①小児期からの貧血共通症状 ②溶血性貧血症状:黄疸、脾腫、ビリルビン系胆石など ③無形成発作:パルボウイルスB19感染を合併すると、ウイルスが赤芽球に感染して破壊するため貧血が増悪する(急性赤芽球癆)。 【合併症】 葉酸欠乏 |
| 検査 | 【血液検査】 溶血共通:間接Bil↑LDH↑K↑ハプトグロビン↓網赤血球↑ 直接Coombs試験(ー):抗体は関係ないためAIHAを否定できる 自己溶血試験:赤血球浸透圧抵抗性減弱を示す 【末梢血塗抹標本】 球状赤血球(正常な赤血球は中央が薄く凹みがあるため透けて見えるが、それがない) |
| 治療 | 学童期以降に脾摘、同時に胆囊摘出術を行うことが多い (易感染性になるため伝染性紅斑の患者には近づかない!) |
発作性夜間ヘモグロビン尿症 PNH:Paroxymal Nocturnal Hemoglobinuria
| 疫学 | 中年以降に好発 |
| 病態 | 血球はGPIアンカーによって繋ぎ止められたCD55やCD59といった補体制御蛋白によって補体からの攻撃(血管内溶血)を免れている。 PNHは後天的に造血肝細胞のPIG-A遺伝子が変異し、GPIアンカーが欠損した結果、補体は血球を攻撃し汎血球減少が起こる(特に赤血球↓)。その時、赤血球からADPが放出されるため血小板凝集が促進され静脈血栓症をきたす。PNHは一部、再生不良性貧血やMDSに相互移行する場合がある。 |
| 症状 | ①早朝の褐色尿:夜間で換気量低下による呼吸性アシドーシスになると補体が活性化されHb尿↑(ワインカラー尿)。感冒後に補体活性化されて褐色尿になることもある。 ②貧血共通症状、易感染性、出血傾向 ③溶血性貧血症状:眼球結膜黄染、黄疸、胆石 ④血栓症状:腹痛(腸間膜静脈血栓)、腎機能低下、男性機能不全 |
| 検査 | 【血液検査】 溶血共通:間接Bil↑LDH↑↑K↑ハプトグロビン↓網赤血球↑ 赤血球AChE活性↓、NAPスコア↓ 【尿検査】 Hb尿、Hbが代謝されヘモジデリン尿(血管内溶血の所見) 【フローサイトメトリ】 CD55陰性・CD59陰性 【赤血球に対する補体感受性試験】 Ham試験(+):血清を塩酸で酸性化すると補体副経路が活性化する 砂糖水試験(+):イオン強度の低い水中では補体がRBCに結合し溶血しやすい |
| 治療 | 溶血時:ハプトグロビン静注(Hb尿は腎障害を起こすため) 貧血症状:赤血球輸血、ステロイド、エクリズマブ(抗補体抗体) 静脈血栓症:血栓症発症時はヘパリン持続点滴、慢性期はワルファリン内服 造血不全:シクロスポリン、抗胸腺細胞グロブリン 根治療法:造血幹細胞移植 |
赤血球増加症(旧多血症)
赤血球増多症はRBC600万/μL以上、Hb18g/dL以上、Ht50%以上が目安
| 疑われる疾患 | 検査 | |
| 絶対的赤血球増加症 (循環赤血球量↑) |
<二次性> ①低酸素状態:高地、メトHb血症、喫煙(好中球↑)、COPD、先天性心疾患、睡眠時無呼吸症候群など ②EPO産生腫瘍:腎細胞癌、肝細胞癌 ③薬剤性:蛋白同化ホルモン、EPO製剤 |
EPO↑ NAP正常 |
| <原発性> 真性赤血球増加症(PV)→骨髄増殖性腫瘍を参照 |
EPO↓ | |
| 相対的赤血球増加症 (循環赤血球量不変) |
①脱水などで血液濃縮状態(循環血漿量↓) ②ストレス多血症:ストレス、高血圧 |
EPO正常 |
止血・凝固系の疾患
出血傾向の鑑別
| 出血T↑ | PT↑ | APTT↑ | その他 | ||
| ①血小板数の低下 【産生障害】 |
再生不良性貧血、急性白血病、PNH、MDS | ||||
| 巨赤芽球性貧血: | |||||
| ①血小板数の低下 【破壊亢進】 |
DIC | ↑ | ↑ | ↑ | |
| TTP | ↑ | 正常 | ADAMTS-13 | ||
| ITP | ↑ | 正常 | 正常 | ピロリ菌 | |
| HUS:先行感染、溶血性貧血、急性腎障害 | ↑ | ||||
| ①血小板数の低下 【異常プール】 |
肝硬変など脾腫を来たす疾患 | ||||
| ②血小板機能の低下 | 血小板無力症 | ↑ | 正常 | 正常 | |
| von Willebrand病 | ↑ | 正常 | ↑ | 鼻出血 | |
| 尿毒症 | ↑ | 正常 | 正常 | ||
| NSAIDs(アスピリンなど) | ↑ | 正常 | 正常 | ||
| ③凝固因子の欠乏 | 血友病 | 正常 | 正常 | ↑ | |
| von Willebrand病 | ↑ | 正常 | ↑ | 鼻出血 | |
| 線溶抑制型DIC | ↑ | ↑ | ↑ | ||
| ビタミンK欠乏症 | 正常 | ↑ | ↑ | ||
| ④線溶系の暴走 | 線溶亢進型DIC | ↑ | ↑ | ↑ | FDP↑ |
| ⑤血管壁の脆弱性 | Osler病 | 正常 | 正常 | 正常 | 鼻出血 |
| IgA血管炎 | 正常 | 正常 | 正常 | 腹痛、関節痛 | |
| 壊血病 | 正常 | 正常 | 正常 |
血小板無力症(Glanzmann病)
| 病態 | 常染色体劣性遺伝により先天的にGPⅡb/Ⅲaに異常があり、血小板凝集能が低下して出血傾向を呈する疾患。 |
| 症状 | 幼少期からの表在性出血傾向(鼻・歯肉・下血といった粘膜出血、紫斑) |
| 検査 | 出血時間↑(PTL数正常、PT・APTT正常) |
| 治療 | 出血に対する対症療法 |
血栓性血小板減少性紫斑病 TTP:Thrombotic thrombocytopenic purpura
| 疫学 | 40歳以上の女性、60歳以上の男性 |
| 病態 | 後天的にvWF切断酵素(ADAMTS13)に対する自己抗体ができ、ADAMTS13の活性低下すると巨大vWFが出現し、血小板血栓が多発する。また、巨大vWFに赤血球がぶつかり破砕赤血球が生じ、溶血性貧血や各種臓器障害をきたす。 |
| 症状 | <五主徴> ①〜③はHUSと共通! ①出血傾向:PLT↓による紫斑など ②溶血性貧血症状: ③乏尿、血尿、尿蛋白:血栓による血流障害から急性腎不全になる ④動揺性の精神神経症状:血栓による血流障害により頭痛、せん妄、痙攣、意識障害など ⑤発熱:原因不明 人生初瀕死:腎障害、精神症状、発熱、貧血、出血傾向 |
| 検査 | 【血液検査】PLT↓出血時間↑、WBC↑、RBC↓、BUN&Cre↑、PT&APTT正常(DICとの鑑別)、ADAMTS-13活性↓ 溶血共通:間接Bil↑LDH↑K↑ハプトグロビン↓網赤血球↑ 【末梢血塗抹標本】破砕赤血球 |
| 治療 | 血漿交換により不要な自己抗体を除去、新鮮凍結血漿(ADAMTS13補充)+ステロイド(自己抗体消失) ※血小板輸血は禁忌(血栓が多発するため) |
免疫性血小板減少性紫斑病 ITP:Immune thrombocytopenic purpura
| 疫学 | 急性型は小児に多い、慢性型は成人女性と高齢者に多い |
| 病態 | 旧 特発性血小板減少性紫斑病。 免疫学的機序で血小板自己抗体が産生され、自己抗体と結合した血小板が脾臓でⅡ型アレルギーにより血小板破壊が亢進して出血傾向をきたす。急性型と慢性型に分類される。Hピロリ菌感染が関与している症例がある。 急性型:先行感染(風疹、麻疹、水痘)を伴ったが多く、6ヶ月以内に治癒する。 慢性型:先行感染がない場合が多く、寛解しにくい。 |
| 症状 | 紫斑(点状出血)、粘膜出血(鼻出血、歯肉出血、性器出血)、脳出血 上記出血により貧血が生じることがある。 【合併症】 AIHA(Evans症候群)、SLE、抗リン脂質抗体症候群など自己免疫異常を合併しやすい |
| 検査 | 【血液検査】PLT↓出血時間↑、PT&APTT正常、抗血小板抗体(PAIgG)↑ 【尿素呼気検査】ピロリ菌感染者のスクリーニング 【骨髄血塗抹標本】巨核球↑ないし正常 |
| 治療 | ①ピロリ菌陽性者はまず除菌療法(陽性者の6割で血小板数が回復する) ②PLT3万未満 or 出血傾向の場合:ステロイド経口投与(4週間) ③ステロイド無効例は脾摘+γグロブリン大量静注(投与で一過性に血小板の寿命を延ばす)→PLT1万未満、頭蓋内出血、広範な紫斑・粘膜出血などの緊急時にも ④抵抗性の場合、TPO受容体作動薬(血小板↑)、リツキシマブ投与。 |
IgA血管炎(アレルギー性紫斑病→シェーンライン・ヘノッホ紫斑病)
膠原病の血管炎を参照。
溶血性尿毒症症候群 HUS:Hemolytic uremic syndrome
HUS発症例では菌が分離されなくてもVero毒素が検出されれば、直ちに保健所長を経由して都道府県知事に届出する。
| 疫学 | 大腸菌によるものは4歳以下の乳幼児に好発 |
| 病態 | TTPに類似する病態だが、HUSは主に腎臓が障害される。 【原発性】腸管出血性大腸菌の産生するvero毒素(志賀毒素)→焼肉などが原因 EHECは腸管上皮細胞に付着して志賀毒素を産生し腸管や血管内皮細胞の損傷を起こす。 【二次性】抗癌薬、免疫抑制薬、経口避妊薬、妊娠・分娩など |
| 症状 | 原発性は3~7日の潜伏期を経て感染症状(発熱、腹痛、下痢→血便)が出現し、 血便が出現した後に、 <三主徴> ①急性腎不全(主):腎臓の微小血管の血管内皮細胞が傷害され、Cre↑、乏尿、血尿、尿蛋白、顔面・下肢の浮腫などが生じる ②血小板減少:傷害された微小血管で血小板血栓を形成し血小板↓して出血傾向となる ③溶血性貧血症状:血小板血栓に赤血球がぶつかり破壊され、動悸、息切れなど生じる ④その他:高血圧、中枢神経症状(10%:けいれん、意識障害など) ※TTPと症状が似ているが、精神症状はほぼない! HUSはハ(破砕赤血球←溶血性貧血)ジ(腎障害)ケ(血小板減少) |
| 検査 | 【便培養】 原発性はEHEC陽性→O157:H7型が最も多く分離される(約70%) 【血液検査】 PLT↓出血時間↑、WBC↑、RBC↓、BUN&Cre↑、PT&APTT正常 溶血共通:間接Bil↑LDH↑K↑ハプトグロビン↓網赤血球↑ ※ADAMS13に対する自己抗体は産生されない! 【末梢血塗抹標本】 破砕赤血球 |
| 治療 | 急性腎不全には透析、貧血には赤血球輸血 (原発性は抗菌薬投与で溶菌し、毒素放出が増加するため抗菌薬投与×) |
von Willebrand病 vWD
| von Willebrand病 | 後天性von Willebrand病 |
|
| 病態 | 常染色体優性遺伝によりvWF因子が欠乏し、第8因子活性低下が起こり血小板の血管内皮下組織への粘着が不良となり出血傾向を示す。 | vWF因子に対する自己抗体の産生、ADAMTS-13によるvWF因子分解促進などによりvWF因子が欠乏する疾患。ASや人工心臓などの基礎疾患が背景にある。 |
| 症状 | 幼少期からの表在性出血傾向(鼻・歯肉・下血といった粘膜出血、紫斑) | 左に同じ? |
| 検査 | 【血液検査】 vWF欠乏による出血時間↑&APTT↑(PLT数・PTは正常)、血小板粘着能↓、リストセチン凝縮能↓ |
左に同じ? |
| 治療 | 第8因子+vWF複合体製剤 軽症ならデスモプレシン(血管内皮細胞から第8因子を血中に放出させる) |
左に同じ? |
血友病 Hemophilia
| 血友病 | 後天性血友病 | |
| 疫学 | 男性のみ(伴性劣性遺伝) | 男女 |
| 病態 | 第8 or 9因子(5:1)が欠乏し、十分なフィブリン血栓が作れず出血傾向を示す。幼少期からの深部出血傾向あり。 | 後天的に第8因子に対する自己抗体が産生され、十分なフィブリン血栓が作れず出血傾向を示す。SLEなど合併しやすい。 |
| 症状 | ①関節内・筋肉内出血 ②有痛性腫脹 |
①筋肉内出血(関節内はない!) ②皮下出血・紫斑 |
| 検査 | 【血液検査】APTT↑ (出血時間・PLT数・PTは正常) |
【血液検査】APTT↑ (出血時間・PLT数・PTは正常) |
| 治療 | 血友病A:第8因子補充+デスモプレシン 血友病B:第9因子補充 緊急止血:第7因子製剤、プロトロンビン |
後天性血友病:ステロイド、効果不十分ならシクロホスファミドなど 緊急止血:左に同じ |
ビタミンK欠乏症
生化学のビタミンを参照
DIC:Disseminated intravascular coagulation(播種性血管内凝固症候群)
| 病態 | 基礎疾患によって高サイトカイン状態となり、血小板や凝固因子が活性化される。その結果、微小血栓ができて虚血により多臓器障害を起こす。また、血小板や凝固因子の消耗+微小血栓を溶かそうと線溶系が亢進し出血傾向を示す。血栓傾向が強いか出血傾向が強いかは場合による(線溶抑制型、線溶均衡型、線溶亢進型)。 <代表的基礎疾患> ①敗血症(線溶抑制型):臓器障害が生じやすく、出血は少ない ②固形悪性腫瘍(線溶均衡型):進行例を除き、臓器障害・出血ともに少ない ③APLなどの急性白血病(線溶亢進型):出血は高度だが、臓器障害は少ない ④その他:急性膵炎、劇症肝炎、外傷や火傷による組織損傷、産科疾患(常位胎盤早期剥離、羊水塞栓)、脱水症など |
| 症状 | 点状紫斑、粘膜出血(PLT減少による) 深部出血(凝固因子減少による) 呼吸困難、意識障害、乏尿、黄疸、ショックなどの臓器虚血症状 |
| 検査 | 【血液検査】 血小板の消費:PLT↓出血時間↑ 凝固因子の活性化:PT&APTTの延長 凝固因子の消費:ATⅢが消費性に減少しTAT↑(トロンビン-アンチトロンビンⅢ複合体) 微小血栓形成:フィブリノゲン↓、末梢血塗抹標本で破砕赤血球 線溶系亢進:プラスミノーゲン↓プラスミン-α2複合体(PIC)↑FDP&D-dimer↑ |
| 治療 | 原疾患の治療+抗凝固療法(ヘパリン、AT製剤、合成プロテアーゼ阻害薬) 適宜、濃厚血小板(PLT5万未満)や新鮮凍結血漿(=凝固因子)を補充 |
造血器腫瘍
【染色法】
| May-Giemsa染色 | 細胞の核や細胞質成分を染色する方法 |
| MPO染色 | 主に好中球の細胞質に含まれるペルオキシダーゼを染色する方法で、に黒褐色 or 黄褐色 or 青色斑点が見えるものが陽性 |
【白血病の鑑別】
| 鑑別点 | |
| AML | 血小板↓、白血病裂孔+、MPO染色+、異常芽球が単一に↑(分化能ー) |
| ALL | 血小板↓、白血病裂孔+、MPO染色ー、一部ALLでPh染色体+、異常芽球が単一に↑ |
| CML | 血小板↑、白血病裂孔ー、各分化段階の血球が見られる(分化能+) |
| CLL | 成熟したリンパ球が腫瘍化、Ph染色体+ |
| ATL | 成熟したリンパ球が腫瘍化、高Ca血症 |
急性白血病
| 急性骨髄性白血病(AML) | 急性リンパ性白血病(ALL) | |
| 病態 | ||
| 症状 | ||
| 検査 | ||
| 治療 |
【急性白血病の化学療法】
| ①寛解導入療法 | 血液学的完全寛解(造血機能回復、末梢血中異常クローン細胞がいない、骨髄の芽球比率5%未満)まで化学療法を行う。適宜支持療法を行う。 ※開始時は腫瘍崩壊症候群を示し、急性腎不全になりやすい。 |
| ②地固め療法 | 分子生物学的寛解(残存する異常クローン細胞がPCRで検出できない)まで化学療法を継続する。 |
| ③維持・強化療法 | 再発を防ぐため毒性を減らした薬剤を継続的に投与する。 Ph(+)ALLはチロシンキナーゼ阻害薬と化学療法を併用する。 |
急性骨髄性白血病 AML
| 疫学 | 50歳以上に好発 |
| 病態 | 造血幹細胞の分化ある段階で分化能を失い異常芽球(白血病細胞)が単一に増殖し、臓器浸潤する疾患(白血病裂孔+)。 そのため、正常血球は全て減少し、骨髄の芽球比率は20%以上になる。 FAB分類によってM0〜M7に分類する。 MDSなどの血液疾患、抗癌剤投与、放射線治療によって発症リスク↑ |
| 症状 | ①汎血球減少症状:易感染性、貧血、出血傾向 ②臓器浸潤症状:肝脾腫・リンパ節腫脹など ③発熱:免疫応答による腫瘍熱 【M3】DICを高率で合併し、出血症状 【M4・M5】歯肉腫脹・皮下結節 【M7】Down症に合併しやすい |
| 検査 | 【①血液検査】WBC↑RBC↓PTL↓ 【②骨髄血塗抹標本】MPO染色+(陽性芽球が3%以上)、芽球比率20%以上 【③フローサイトメトリ】顆粒球系はCD13・CD33+、単球系はCD14+ 【④染色体検査】下記のFAB分類参照 |
| 治療 | 【M3】 分化誘導療法:ATRA=全トランス型レチノイン酸(無効時は亜ヒ酸)+化学療法(アントラサイクリン系+シタラビン:APL細胞殺傷してATRA症候群予防) 【M3以外】 化学療法(アントラサイクリン系+シタラビン)→造血幹細胞移植 |
【FAB分類のAMLの名称】
| 型 | 特徴 | |
| M0 | 最未分化型 | 骨髄芽球への分化途中で腫瘍化。AMLの数%だが難治性。 【骨髄血塗抹標本】CD13・CD33+(骨髄球系マーカー陽性)。AMLなのにMPO染色ー |
| M1 | 未分化型 | 好中球への分化途中で腫瘍化。AMLの10%で、予後比較的良好。 【骨髄血塗抹標本】特異的エステラーゼ+ |
| M2 | 分化型 | 好中球への分化途中で腫瘍化。AMLの20%。比較的予後良好。 【染色体異常】M2はt(8;21)(蜜はハニー) 【骨髄血塗抹標本】特異的エステラーゼ+ |
| M3 | 前骨髄球性 (APL) |
好中球への分化途中で腫瘍化。AMLの15%で、予後良好。 【染色体異常】t(15;17)(一個だけ予後いーな)、この異常により異常なレチノイン酸受容体が形成される。 【骨髄血塗抹標本】特異的エステラーゼ+、MPO染色でアズール顆粒+、それが融合したAuer小体が多数あり、それが含まれるfaggot細胞が見られる。アズール顆粒内には第3凝固因子を多く含むため、高率に線溶亢進型DICを合併してフィブリノゲン↓FDP↑となる(強い出血傾向)。 |
| M4 | 骨髄単球性 (AMMoL) |
単球&顆粒球への分化途中で腫瘍化。AMLの20% 【骨髄血塗抹標本】特異的&非特異的エステラーゼ+(両方とも陽性) |
| M5 | 単球性 (AMoL) |
単球への分化途中で腫瘍化。AMLの5%。 【骨髄血塗抹標本】非特異的エステラーゼ+(特異的はー) |
| M6 | 赤芽球性 | 赤血球&顆粒球への分化途中で腫瘍化。AMLの5%未満だが難治性。 【骨髄血塗抹標本】骨髄中赤芽球50%以上、PAS染色で巨大赤芽球+ |
| M7 | 巨核芽球性 | 血小板への分化途中で腫瘍化。AMLの5%未満。 Down症に好発する。また、M7の30%以上に骨髄線維症が合併する。 【骨髄血塗抹標本】CD41・CD61+(単球系)、PPO(血小板ペルオキシダーゼ)染色+、AMLなのにMPO染色ー |
急性リンパ性白血病 ALL
| 疫学 | 小児(Ph-が多い)と高齢者の二峰性 |
| 病態 | 造血幹細胞の分化ある段階で分化能を失い異常芽球(白血病細胞)が単一に増殖し、臓器浸潤する疾患。そのため、正常血球は全て減少し、骨髄の芽球比率は20%以上になる。B細胞系のALLが80%以上。 【予後不良因子】 ①年齢:30歳以上 ②WBC数:3万以上 ③染色体:t(9;22)、t(4;11) 一部ALLでPh染色体陽性t(9;22)となり予後不良となる(CMLのPh染色体と切断点が異なるため生成蛋白も異なるためTKIだけではCML同様の長期生存は望めない)。 |
| 症状 | ①汎血球減少症状:易感染性、貧血、出血傾向 ②臓器浸潤症状:肝脾腫・リンパ節腫脹など ③発熱:免疫応答による腫瘍熱 Down症候群に合併しやすい AMLに比べて中枢神経浸潤しやすい(頭痛、嘔吐、麻痺など) |
| 検査 | 【①血液検査】WBC↑↑、代償的にRBC↓PTL↓ 【②骨髄血塗抹標本】 ・MPO染色ー(3%未満)、芽球比率20%以上 ・TdT陽性(Terminal deoxynucleotidyl transferase):T細胞受容体やIgの再構成に必要な酵素(急性リンパ性白血病のマーカー) 【③フローサイトメトリ】 T細胞系はCD2/3/5/7+、B細胞系はCD10/19/20+(T細胞系は1桁、B細胞系は2桁) |
| 治療 | ①化学療法:ビンクリスチン+アントラサイクリン系+L-アスパラギナーゼ ②プレドニゾロン: ③小児では中枢神経浸潤予防のため必ずメトトレキサート+シタラビン大量髄注 ④Ph+の場合:上記の化学療法+イマチニブ→第一寛解期に造血幹細胞移植 ※小児は予後良好であり、化学療法だけでもかなりの症例が根治する。 |
慢性骨髄性白血病(CML)
| 疫学 | 50〜60代に好発 |
| 病態 | 9番染色体のABL遺伝子と22番染色体のBCR遺伝子が転座により融合してBCR-ABL融合遺伝子が形成され、造血幹細胞にPh染色体t(9;22)が発生し(マンコツにクンニ)、分化能を保ったまま各段階の顆粒球が増殖する(白血病裂孔ー)。無治療だとCML確定診断から3〜4年後に急性転化することが多い。 【急性転化】 CMLの経過中に造血幹細胞が分化能を失い、急性白血病の症状が出現する(MPO染色陽性率により、AML(70%)、ALL(30%)に転化かを判断)。予後は不良。 |
| 症状 | 無症状(健診で白血球増多を指摘) 食欲不振により倦怠感や体重減少、髄外造血により脾腫(ほぼ必発) 著名な肝脾腫があれば腹部膨満感 |
| 検査 | 【血液検査】 WBC↑(好塩基球↑が特徴)、RBC↓、PLT↑、白血球崩壊によってLDH↑尿酸↑ビタミンB12↑(白血球内にVB12結合蛋白が存在しており、血球の破壊により放出される)、NAPスコア↓ (急性転化した場合)WBC↑(白血病裂孔+)、RBC&PLT↓、NAPスコア↑ 【骨髄生検】 顆粒球系の高度な過形成、(急性転化した場合)dry tap |
| 治療 | 【薬物療法】チロシンキナーゼ阻害薬のイマチニブ、ニロチニブ、ボスチニブ、ダサチニブ、ボナチニブ(根治はできず内服継続が必要)→イマチの煮干しダサい 【薬物療法抵抗性】造血幹細胞移植、化学療法 |
類白血病反応
| 病態 | 癌転移、重症感染症、骨髄線維症、薬剤中毒などの基礎疾患に対する反応として血液所見が白血病(特にCML)に酷似する病態。 |
| 診断 | WBC:30000以上、または骨髄球以前の幼若白血球の末梢血の出現 |
| 鑑別 | CMLと異なりPh染色体(ー)、好塩基球の増加(ー) |
| 治療 | 基礎疾患の治療 |
慢性リンパ性白血病(CLL)
| 疫学 | 60歳以上に好発、全白血病の3%と日本では少ない |
| 病態 | CD5陽性の成熟小型B細胞が緩徐に増殖し、リンパ節や脾臓に浸潤する(B細胞性なのCD5陽性)。一般的に予後良好だが、進行すると感染症や白血病増悪で死亡することがある。 |
| 症状 | 無症状(健診で白血球増多を指摘) 進行すると易感染性、リンパ節腫脹、肝脾腫 腫瘍B細胞が異常なIgを作るため自己免疫疾患を合併しやすく(AIHA、ITPなど)、正常な免疫グロブリン産生が抑えられて低γグロブリン血症となる。 |
| 検査 | 【血液検査】WBC(リンパ球)↑、CD5+成熟小型Bリンパ球↑、低γグロブリン血症 【ツベルクリン反応】陰転化(細胞性免疫低下→悪性腫瘍を合併しやすい) 【フローサイトメトリ】CD5/19/20/23などのリンパ球表面抗原陽性(B細胞系なのにT細胞系マーカーCD5が出現する謎!!がCLLの特徴) |
| 治療 | 無症状:経過観察(抗癌剤抵抗性のため) 症状がある場合:多剤併用化学療法(フルダラビン)、BTK阻害薬(イブルチニブ) |
成人T細胞白血病(ATL)・成人T細胞リンパ腫(ATLL)
ウイルス学のHTLV-1を参照。
骨髄増殖性腫瘍 MPN:Myeloproliferative neoplasms
造血幹細胞がクローン性に増殖し、成熟血球が増加する病態。
造血サイトカインの受容体からのシグナル → JAK2が活性化 → G-CSF↑EPO↑TPO↑
| 原因遺伝子 | その他 | |
| ①慢性骨髄性白血病(CML) | フィラデルフィア染色体形成 | 巨脾 |
| ②原発性骨髄線維症(PMF) | JAK2遺伝子変異(50%に認める) | 巨脾 |
| ③本態性血小板血症(ET) | JAK2遺伝子変異(50%に認める) | 巨脾なし |
| ④真性赤血球増加症(PV) | JAK2遺伝子変異(95%に認める) | 巨脾 |
原発性骨髄線維症 PMF:Primary myelofibrosis
| 疫学 | 60歳代に好発 |
| 病態 | 約50%にJAK2遺伝子変異があり、巨核球が増殖する変異が起こり、その巨核球から放出されるサイトカインによって線維芽細胞が増加し、二次的に骨髄が線維化する。そのため、髄外造血が起こり異常な血球(涙滴赤血球、巨大血小板、白赤芽球症=髄外造血によって赤芽球・骨髄芽球が末梢血に出現)が生じて様々な症状を生じる。 |
| 症状 | ①低酸素症状:貧血のため ②左季肋部痛・腹部膨満感:肝脾腫、特に巨大脾腫は必発する |
| 検査 | 【血液検査】 WBC↑好酸球↑幼若細胞(+)、NAPスコア↑、RBC↓、PTL↑ 血球崩壊により尿酸↑LDH↑ビタミンB12↑ 【末梢血塗抹標本】 涙滴赤血球(線維化でぴえん)、巨大血小板・赤芽球・骨髄芽球(白赤芽球症:leukoerythroblastosis) 【骨髄穿刺】dry tapで吸引不可 →【骨髄生検】鍍銀染色で線維が黒く染色 |
| 治療 | 低リスク群:経過観察 高リスク群:根治として同種造血幹細胞移植 対症療法:貧血には蛋白同化ステロイド、脾腫にはJAK阻害薬 |
本態性血小板血症 ET:Essential thrombocythemia
| 疫学 | 中高年に好発 |
| 病態 | 約50%にJAK2遺伝子変異があり(他はCALR変異またはMPL変異)、巨核球の前駆細胞に異常増殖する変異が起こり、血小板が増加する疾患。血小板増加により血栓症が生じ、異常血小板により出血傾向を示す場合もある。予後は比較的良好のため、血栓症と出血の予防に注意する。 |
| 症状 | 慢性経過でほぼ無症状 軽〜中等度の脾腫、点状紫斑や粘膜出血など出血傾向 |
| 検査 | 【血液検査】 WBCやや↑、PLT↑(約100万)、NAPスコア↑、血球崩壊により尿酸↑LDH↑ビタミンB12↑ |
| 治療 | 60歳未満かつ血栓症なし:経過観察 60歳以上または血栓症あり:低用量アスピリン+ヒドロキシカルバミド(ハイドロキシウレア) |
真性赤血球増加症(真性多血症) PV:Polycythemia vera
| 疫学 | 中高年 |
| 病態 | ほぼ100%にJAK2遺伝子変異があり、赤血球を中心に汎血球増加する変異が起こる疾患。 長期経過後に骨髄線維症やAMLへ転化することがある。 |
| 症状 | ①顔面紅潮(赤ら顔):RBC↑循環血液量↑のため ②頭痛、耳鳴、めまい:RBC↑により血液粘稠性↑して血流低下による低酸素状態 ③血栓症状(DVTなど):PLT↑のため ④皮膚掻痒感:好塩基球↑によるヒスタミン分泌↑ ⑤肝脾腫:RBC処理↑により脾腫、髄外造血により肝臓腫 |
| 検査 | 【血液検査】 RBC↑(平均値の25%以上)、Hb↑(男性16.5以上、女性16以上)、代償的にEPO↓ WBC↑、PTL↑、NAPスコア↑、血球崩壊により尿酸↑LDH↑ビタミンB12↑ 【骨髄生検】 赤芽球、顆粒球、巨核球の3血球系統の過形成(特に赤芽球系↑) |
| 治療 | 60歳未満かつ血栓症の既往なし:低用量アスピリン(血栓症予防)+瀉血(RBC↓) 60歳以上または血栓症の既往あり:低用量アスピリン+瀉血+ヒドロキシカルバミド(ハイドロキシウレア)、不応例はJAK2阻害薬も行われる。 |
骨髄異形成症候群 MDS:Myelodysplastic syndrome
| 疫学 | 60〜70歳に好発、5q-症候群は日本では非常に稀 |
| 病態 | 半数以上に、5q-、7q-、20q-、+8などの染色体異常(約50%)により、造血幹細胞から正常な分化をせずに異常な芽球がつくられ、あまりに異常な芽球は骨髄内で破壊され末梢血に出ず(無効造血)、汎血球減少が起こる。また、分化能を失い芽球比率が20%以上になるとAMLに移行する(前AML状態)。 【MDSの予後予測スコアリングシステム(IPSS)】 骨髄芽球比率、染色体異常、血球減少を用いて予後を予測し、リスク群を分類 低リスク群:数年以上の余命が期待できる 高リスク群:早期に白血病化して1〜2年以内に死に至る |
| 症状 | ①汎血球減少症状:易感染性、貧血、出血傾向 無症状のことも多く健診で発見される。 |
| 検査 | 【血液検査】 汎血球↓(無効造血)、代償的にEPO↑ 【骨髄血塗抹標本】 骨髄正〜過形成、様々な血球異形成(多核の赤芽球、顆粒の乏しい骨髄球)、芽球増加+比率20%未満(※正常骨髄の芽球比率は5%未満)、RBCは正球性と大球性の二相性 |
| 治療 | 【低リスク群】症状なければ経過観察、貧血には赤血球輸血、血小板減少には血小板輸血。5q欠損はレナリドミドが奏功し、予後良好となる。 【高リスク群】造血幹細胞移植、移植困難な場合はアザシチジン投与 |
治療関連性白血病(二次性白血病)
| 病態 | 悪性腫瘍に対して化学療法や放射線照射を行った後に生じる白血病(多くはAML)。特にアルキル化薬とトポイソメラーゼⅡ阻害薬は本症を起こしやすい。化学療法抵抗性で予後は不良。※造血幹細胞移植後は口腔癌、食道癌、皮膚癌が多い。 |
| 症状 | 治療5〜6年後にMDS→AMLの症状 |
| 検査 | AMLに準じる |
| 治療 | AMLに準じる |
腫瘍崩壊症候群
| 病態 | 白血球過剰時の化学療法によって細胞内の物質が血中に逸脱し、高K血症・高P血症・高尿酸血症となり、崩壊した細胞により血流が途絶えると組織酸欠となり乳酸アシドーシスを来たす。高P血症では血中Caと結合して骨組織に取り込まれ低Ca血症となる。 |
| 症状 | 化学療法開始後に生じ、高尿酸血症により急性腎不全・尿路結石、高K血症により不整脈、低Ca血症によるテタニーなどを来たす。 |
| 検査 | 【血液検査】K↑、P↑、Ca↓、尿酸↑(腫瘍細胞破壊による核酸放出のため) |
| 治療 | 高尿酸血症対策:大量輸液、尿酸生成抑制薬(フェブキソスタット)、尿酸分解酵素薬(ラスブリカーゼ)、尿アルカリ化薬 |
多発性骨髄腫 MM:Multiple myeloma
| 疫学 | 50歳以上に好発 |
| 病態 | 末梢の形質細胞が骨髄や髄外リンパ節に移動して単クローン性に腫瘍化し、腫瘍細胞が異常なモノクローナル抗体(M蛋白:IgG型>IgA型>BJ型)を産生し、骨髄腫関連障害(CRABO:クラブ)を起こす疾患。Bence-Jones蛋白は免疫グロブリン軽鎖のみからなる蛋白。 【予後不良因子】 血中Alb↓、血中β2MG↑でステージングを行う |
| 症状 | ①Calcium:高Ca血症:腫瘍細胞が破骨細胞活性化因子(OAF)を分泌し、破骨細胞が骨吸収を促進して高Ca血症を起こす。高度になると意識障害、腎障害をきたす。 ②Renal:腎機能障害:BJ蛋白のみ糸球体通過(アミロイドも)し、尿細管に沈着し腎機能を低下させる。また、尿細管性アシドーシスも生じる。 ③Anemia :貧血、Amiroidosis:アミロイドーシス:骨髄が異常クローンにより占拠され造血↓する。Igの軽鎖はアミロイドに変性する(=ALアミロイドーシス)。 ※脾腫は呈さない(脾臓に浸潤したり髄外造血したりしないため) ④Bone:骨折:OAFにより骨吸収が促進され、腰背部痛などの骨痛(脊椎圧迫骨折)、骨打ち抜き像などの骨融解が多発する。 ⑤Other(感染症、過粘稠度症候群):異常抗体しかできないため易感染性となる。M蛋白が赤血球連鎖を形成し血液の粘稠性が↑し過粘稠度症候群を来たす(頭痛、めまい、悪心嘔吐、出血傾向など)。 |
| 検査 | 【血液検査】M蛋白↑により総蛋白↑(BJ型+は尿中排泄されるため総蛋白不変)、Ca↑、正常なグロブリン↓、総蛋白-Alb乖離、腎機能低下でBUN・Cre↑、RBC↓、赤血球連鎖により赤沈↑ 【尿検査】BJ型の場合はBJ蛋白+ 【電気泳動】 γグロブリン↑によりMピーク形成。免疫電気泳動でM蛋白のM-bow(分厚い弓)確認 【骨髄血塗抹標本】 核が偏在+核周囲明庭+細胞質の広い異型形質細胞を確認 ※骨髄穿刺は上後腸骨棘で行う(病的骨折が生じる危険性のため胸骨は禁忌) 【画像検査】 頭蓋骨単純X線:骨打ち抜き像(punched-out-lesion)(溶骨病変) CT:単純CTは腰椎病変見るのに良いが、造影は腎機能悪化させるため原則禁忌! |
| 治療 | 無症候の場合:経過観察 症候があり65歳未満の場合:自家造血幹細胞移植+大量化学療法(メルファラン) 症候があり65歳以上または重症臓器障害がある場合:以下の薬物療法 ①プロテアソーム阻害薬:ボルテゾミブ、カーフィルゾミブ、イクサゾミブ →プロテアソームを阻害して腫瘍形質細胞をアポトーシスさせる ②免疫調整薬:サリドマイド、レナリドミド、ポマリドマイド、ステロイド ③抗CD38抗体:ダラツズマブ ④ヒストン脱アセチル化酵素阻害薬:パノビノスタット |
MGUS(エムガス)
| 疫学 | 高齢者に好発 |
| 病態 | モノクローナル性γグロブリン増加でM蛋白が認められるが、無症状で予後良好な疾患。 ただし、10年で約10%はMMへ移行する。 |
| 症状 | 無症状 |
| 検査 | 【血液検査】M蛋白3g/dL未満、他のIg値は正常(MMでは低下) 【免疫電気泳動】M蛋白のM-bow(分厚い弓)確認 |
| 治療 | 経過観察 |
悪性リンパ腫 ML:Malignant Lymphoma
| リンパ性白血病 | 異常クローン細胞が骨髄や末梢血で増殖する→臓器浸潤 |
| 悪性リンパ腫 | 異常クローン細胞がリンパ節や脾臓で増殖する→末梢血などに浸潤(白血化) |
| 疫学 | Hodgkinリンパ腫(5%)、非Hodgkinリンパ腫(95%) Hodgkinリンパ腫は20歳代と60歳代の2峰性、非Hodgkinリンパ腫は60代に好発 |
| 病態 | リンパ系細胞が腫瘍化し、リンパ組織で腫瘤を形成する疾患。 <非Hodgkinリンパ腫> ①B細胞系:びまん性大細胞型B細胞リンパ腫>濾胞性リンパ腫>MALTリンパ腫>マントルリンパ腫>Burkittリンパ腫など ※非Hodgkinの予後因子(国際予後指標)はALPSE:Age、血清LD、PS、Stage、Extranodal sites(節外病変数) ※中枢神経系原発悪性リンパ腫のほとんどが非Hodgkinリンパ腫 ②T細胞系:ATLL、菌状息肉症、セザリー症候群など 【Ann Arbor病期分類】 Ⅰ期:横隔膜の上部の1領域にリンパ節腫脹あり(B症状なしはⅠA、ありはⅠB) Ⅱ期:横隔膜の上部の複数領域にリンパ節腫脹あり(B症状なしはⅡA、ありはⅡB) Ⅲ期:横隔膜の上下両方にリンパ節腫脹あり+脾に浸潤 Ⅳ期:横隔膜の上下両方にリンパ節腫脹あり+肝や骨髄など非リンパ臓器に浸潤 |
| 症状 | ①A症状:全身症状なし ①B症状:38℃以上の発熱、盗汗、半年以内に10%以上の体重減少(異常クローン細胞が放出するサイトカインによる症状) ※Hodgkinリンパ腫では波状熱が見られる(Pel-Ebstein熱) ②リンパ節腫脹・脾腫:腫瘍細胞の浸潤。初発症状として頸部リンパ節腫脹が多い |
| 検査 | 【血液検査】LDH↑、sIL-2受容体↑、炎症によるCRP&赤沈↑ 【リンパ節生検】細胞表面抗原CD20+の場合がある 【画像検査】CT 、MRI、FDG-PET 心エコー:ドキソルビシンは心毒性があり、心機能の評価を行う 【骨髄穿刺】骨髄浸潤を確認【腰椎穿刺】髄膜浸潤を確認【内視鏡】腸管浸潤を確認 |
| 治療 | Hodgkinリンパ腫:ABVD療法(ⅠA・ⅡAの場合は放射線療法併用) 非Hodgkinリンパ腫:CHOP療法、CD20+の場合はリツキシマブ併用 |
※ABVD療法:アドリアマイシン(=ドキソルビシン)、ブレオマイシン、ビンブラスチン、ダカルバジン
※CHOP療法:シクロホスファミド、ドキソルビシン、ビンクリスチン(クリクリビンビンCHOP!)、プレドニゾロン
【代表的な悪性リンパ腫】
| びまん性大細胞型B細胞リンパ腫 (DLBCL) |
悪性リンパ腫で最多(30%)。中枢神経系原発悪性リンパ腫の約90%。 t(3;14)の染色体異常を持ち、濾胞構造が消失するB細胞腫瘍(大家族は妻子持ち)。B細胞系のCD10、CD20が発現。化学療法により半数以上が治癒可能。 |
| 濾胞性リンパ腫 | 悪性リンパ腫の約15%。t(14;18)の染色体異常を持ち、bcl-2過剰発現により濾胞構造が多数存在するB細胞腫瘍(ロッホーと叫んでる医師は嫌)。B細胞系のCD10、CD20が発現。進行期でも無症状なら経過観察を考慮して良い。 |
| マントルリンパ腫 | t(11;14)の染色体異常を持ち、消化管に浸潤しやすい特徴を持つB細胞腫瘍(マンマン好きないい医師)。B細胞系なのにCD5+! |
| MALTリンパ腫 | t(11;18)の染色体異常を持ち、MALTがある消化管が初発でHピロリ菌が関与するB細胞腫瘍。胃MALTでは除菌で60%以上が治癒する。 |
| Burkittリンパ腫 | t(8;14)の染色体異常を持ち、腹部膨満感や腫瘍崩壊症候群を生じやすい特徴を持つ高悪性度のB細胞腫瘍(バーきっと廃止)。一部EBウイルスが関与。B細胞系のCD10、CD20が発現。適切な治療で高率に治癒が期待できる。 |
| Hodgkinリンパ腫 | 悪性リンパ腫の約5%。Hodgkin細胞やReed-sternberg細胞(ふくろうの目あり)を持つ起源不明の腫瘍で、隣接リンパ節に連続性に進展する。白血化は稀で、化学療法感受性も高く予後は比較的良好。 |
| 菌状息肉症 | 皮膚原発の低悪性度のCD4+T細胞悪性リンパ腫。詳細は皮膚科を参照。 |
| セザリー症候群 | 末梢血にセザリー細胞が出現するCD4+T細胞悪性リンパ腫。現在は菌状息肉症が白血化した疾患と考えられている。詳細は皮膚科を参照。 |
| 中枢神経原発悪性リンパ腫 | 眼球内リンパ腫(眼の見えづらさ)が初発症状の一つ(数%) 治療はメトトレキサート大量療法+放射線全脳照射 |
原発性マクログロブリン血症
マクログロブリン=5量体として存在するIgM
| 疫学 | 60歳以上の男性に多い |
| 病態 | 形質細胞に近い成熟B細胞が腫瘍性にリンパ節、肝臓、脾臓などリンパ組織で主に増殖し、モノクローナル性IgM(5量体)を大量に産生する疾患(形質細胞ではなくB細胞!だから悪性リンパ腫の一つに分類される)。赤血球連銭形成により血清の相対粘稠度が上昇してしばしば過粘稠度症候群をきたす(同じ量であればIgM>IgA>IgGの順に起きやすい)。 |
| 症状 | ①悪性リンパ腫類似症状:リンパ節腫脹、肝脾腫、貧血など ②過粘稠度症候群による循環不全症状:眼底網膜静脈怒張による視力障害、酸欠による頭痛・めまい・悪心嘔吐、赤血球連銭が凝固を阻害し出血傾向、IgM著増によるクリオグロブリン上昇のためレイノー症状 |
| 検査 | 【血液検査】リンパ球様細胞↑によりWBC↑、RBC&PLT↓、IgM↑により総蛋白↑ 【電気泳動】モノクローナル性IgM↑によるM蛋白+ 【眼底検査】網膜静脈のソーセージ様変化、眼底出血 |
| 治療 | MMに準じた化学療法、過粘稠度症候群には血漿交換 |
【MMとの比較】
| 多発性骨髄腫 | マクログロブリン血症 | |
| 主な病態 | 骨髄病変が主 | リンパ組織の病変が主 |
| 赤血球連銭形成 | + | +++ |
| 過粘稠度症候群 | + | +++ |
| 眼底変化 | + | +++ |
| 出血傾向、肝脾腫 | 少ない | 多い |
| M蛋白 | IgG・A・D・E、BJP | IgM |
| 腎障害 | 多い | 少ない |
血球貪食症候群(血球貪食性リンパ組織球症:HLH)
| 病態 | CTLやNK細胞に異常が起こり、病原体を排除できない状態を打開するためT細胞が過剰にサイトカインを分泌する。その結果、マクロファージが活性化して血球まで貪食する疾患。 <原因疾患> ①ウイルス感染:特にEBウイルス、SFTSが重要 ②悪性リンパ腫:特に骨髄に浸潤している場合 ③膠原病:特に成人Still病など |
| 症状 | ①高サイトカイン症状:持続する発熱、DICなど ②脾腫、リンパ節腫脹:マクロファージ増加のため ③溶血性貧血、紫斑:組織球が血液細胞を貪食するため |
| 検査 | 【血液検査】汎血球↓、フェリチン↑、LDH↑、sIL-2R↑、溶血や肝障害で間接Bil↑、肝機能障害によるAST↑ALT↑、TG↑(TNF-αのLPL抑制作用による) 【骨髄血塗抹標本・生検】 骨髄穿刺・リンパ節・脾臓生検し、マクロファージによる血球貪食像を確認 |
| 治療 | ①原因疾患の治療+ステロイド+シクロスポリン ②重症例では血漿交換でサイトカイン除去 |
無顆粒球症 Agranulocytosis
| 病態 | 好中球が500/μL以下に著減した状態。 原因は抗甲状腺薬、抗菌薬、抗癌薬、感染症、放射線などがある。 |
| 症状 | 感染症症状:発熱、咽頭・扁桃に白苔を伴う潰瘍、咽頭痛 |
| 検査 | 【血液検査】顆粒球↓ |
| 治療 | 原因薬剤があれば中止 発熱時は抗菌薬投与、骨髄抑制による場合はG-CSF投与 |
亜急性壊死性リンパ節炎(菊池病)
| 疫学 | 若年者に多い |
| 病態 | 主に若年者にみられる圧痛を伴うリンパ節腫大をきたす良性疾患 |
| 症状 | ①発熱などの全身症状 ②頸部リンパ節腫大 |
| 検査 | 【頸部リンパ節生検】 傍皮質を中心に境界の比較的明瞭な壊死とマクロファージの浸潤 |
| 治療 | 通常は数週間で症状が自然消退する |
造血幹細胞移植 Hematopoietic stem cell transplantation
| 自己造血幹細胞移植(主にauto-PBSCT) | 同種造血幹細胞移植 | |
| 適応 | 多発性骨髄腫、悪性リンパ腫 (抗癌剤が効く腫瘍) |
白血病、悪性リンパ腫、PNH、原発性免疫不全症、再生不良性貧血、MDS (抗癌剤だけでは治らない腫瘍→GVL効果によって腫瘍細胞を排除!) |
| 詳細 | 化学療法で腫瘍細胞を減らした後、G-CSF投与を投与し末梢血に動員された造血幹細胞をアフェレーシスにより採取して保存。その後、大量化学療法を行った後、移植を行う。原則65歳未満。 | 大量化学療法+全身放射線療法で腫瘍細胞を減らした後、他人の造血幹細胞(骨髄 25%・末梢血幹細胞 25%・臍帯血 50%)を移植する。フル移植は55歳未満だが、ミニ移植により70歳くらいまで移植可能となった。 |
| 問題点 | GVHDが起こらないため、採取した自己造血幹細胞中に異常クローン細胞が混在していれば再発する。 | 移植後に感染症、GVHDなどの合併症により致死的となる。末梢血移植は慢性GVHDが重症化しやすい。臍帯血移植は生着に時間がかかる&生着不全が10%に起こる。 |
【HLAの組み合わせ】
| 1つの細胞表面に発現するHLAの種類 | |
| MHCクラスⅠ | 父方のアレル α鎖(HLA-A/B/C遺伝子から発現したクラスⅠ分子)=3種類 母方のアレル 同上で3種類 1つの細胞表面に6種類のクラスⅠ分子が存在する。 |
| MHCクラスⅡ | 父方のアレル α鎖(DP-A,DQ-A,DR-A遺伝子から発現したクラスⅡ分子)×β鎖(DP-B,DQ-B,DR-B遺伝子から発現したクラスⅡ分子)=6種類 母方のアレル 同上で6種類 1つの細胞表面に12種類のクラスⅡ分子が存在する。 ※クラスⅡの方が種類が多く結合できるペプチドの数も多い⇨CD4+T細胞を活性化させる必要があるため! |
| 移植時 | 免疫応答に重要な役割を果たすのはA,B,C,DRの4座で、1つの細胞に8種類の抗原を持つことになる。この抗原が一致するほど生着不全やGVHDが少ない。 ①HLA適合血縁者(兄弟でHLAが一致する確率は25%) ②HLA適合非血縁者(骨髄バンクドナーは登録から移植まで約4ヶ月必要) ③HLA1抗原不適合血縁者 ④臍帯血移植ドナー(2抗原不適合でもOK) ⑤HLA半合致ドナー(2〜3抗原不適合でもOK、半合致する確率は親子100%) ※CPAは移植後早期増殖性T細胞を選択的に殺傷し、ハプロ移植が可能となった! |
【同種造血幹細胞移植の流れ】
| 寛解導入 | 化学療法+α |
| 前処置 | 無菌室で超大量化学療法(シクロホスファミド)+全身放射線照射(TBI)により自己造血を破壊。GVHD予防のためタクロリムス+ミコフェノール酸モフェチル投与。 |
| 移植 | ドナー造血幹細胞を経静脈的に輸注 |
| 生着前 | 抗癌剤の毒性や感染症に注意 |
| 生着後 | GVHDや感染症に注意 |
| 退院 | 順調にいけば、移植後2〜3ヶ月で退院 |
【移植後の感染症対策】
| 起因病原体 | 病名 | 予防法 |
| 細菌 | 敗血症 | レボフロキサシン |
| CMV(移植後30〜90日) | CMV肺炎・胃炎・腸炎 | |
| HHV-6 | ヘルペス脳炎 | ホスカルネットNa |
| アスペルギルス | 侵襲性肺アスペルギルス症 | ミカファンギン |
| カンジダ | カンジダ血症 | ミカファンギン、フルコナゾール |
| HSV(移植後2週間まで) | 口内炎 | アシクロビル |
| アデノウイルス | ||
| VZV(移植6ヵ月後から) | 帯状疱疹 | |
| ニューモシスチス | ニューモシスチス肺炎 |
GVHD(Graft vs host disease):ドナーリンパ球による臓器障害
※GVL(Graft vs leukemia effect):ドナーリンパ球により残存異常クローン細胞を排除する効果
| 急性GVHD(皮疹・黄疸・下痢!) | 慢性GVHD(移植後100日以降) | |
| 皮膚 | 掻痒を伴う紅斑・小丘疹 重症化すると紅皮症・TEN様皮疹 |
多形皮膚萎縮、口腔内・眼の乾燥 強皮症様病変、扁平苔癬様病変が特徴 |
| 肝臓 | T-bil上昇→肝機能障害 | 肝機能障害 |
| 腸管 | 嘔気、水様性下痢(最多) →悪化すると腹痛、血便 |
|
| 肺 | ー | 閉塞性細気管支炎 |
発熱性好中球減少症 FN:Febrile Neutropenia
| 病態 | 抗癌剤使用、造血幹細胞移植時に好中球数500/μL未満となり、発熱(37.5℃以上)が生じ、重篤な細菌感染症が疑われる病態。中心静脈カテーテルが感染源となる場合が多い。 |
| 症状 | 発熱 |
| 検査 | 【血液培養2セット】 約50%に黄色ブドウ球菌、緑膿菌、カンジダ、アスペルギルスが認められる。 緑膿菌はエンドトキシンを産生するため、敗血症性ショックを誘発する可能性がある |
| 治療 | 高リスク:抗緑膿菌活性のある抗菌薬、バンコマイシン、G-CSF投与 低リスク:LVFX、CPF |


コメント